

In this we will cover this msc bioinformatics unit 3 comparative and finctional genomics of paper code 802. The syllabus encompasses a comprehensive introduction to proteomics, detailing essential foundational methods such as protein isolation, purification, and diverse sequencing strategies. We will delve into specific analytical techniques, including 2D gel electrophoresis, chromatography, sequencing by Edman degradation, mass spectrometry, and the application of endopeptidases. Furthermore, the unit explores Serial Analysis of Gene Expression (SAGE), the underlying mechanisms and practical tools for gene prediction, and concludes with an examination of protein-protein interaction databases and software tools.
Introduction to Proteomics
The word “proteome” represents the complete protein pool of an organism encoded by the genome. In broader terms, proteomics is defined as the total protein content of a cell or that of an organism. Proteomics helps in understanding alterations in protein expression during different stages of the life cycle or under stress conditions. Likewise, proteomics helps in understanding the structure and function of different proteins as well as protein–protein interactions of an organism. A minor defect in either protein structure, its function, or alteration in expression pattern can be easily detected using proteomics studies. This is important with regard to drug development and understanding various biological processes, as proteins are the most favorable targets for many drugs. Proteomics as a whole can be divided into three kinds as described below:
Functional proteomics
Structural proteomics
Differential proteomics
Techniques Involved in Proteomics Study
Some of the very basic analytical techniques are used as major proteomic tools for studying the proteome of an organism. The initial step in all proteomic studies is the separation of a mixture of proteins. This can be carried out using the Two-Dimensional Gel Electrophoresis technique, in which proteins are first separated based on their individual charges in the first dimension. The gel is then turned 90 degrees from its initial position to separate proteins based on differences in their size. This separation occurs in the second dimension, hence the name 2D. The spots obtained in 2D electrophoresis are excised and further subjected to mass spectrometric analysis of each protein present in the mixture.
Apart from charge and size, there are a number of other intrinsic properties of proteins that can be employed for their separation and detection. One of these techniques is Field Flow Fractionation (FFF), which separates proteins based on their mobility in the presence of an applied field. The difference in mobility may be attributed to different sizes and masses of proteins. The applied field can be of many types such as electrical, gravitational, or centrifugal. This technique helps in determining different components in a protein mixture, different conformations of proteins, their interaction with other proteins, as well as interactions with organic molecules such as drugs.
Steps in Proteomic Analysis
The following steps are involved in the analysis of the proteome of an organism:
- Purification of proteins: This step involves extraction of protein samples from whole cells, tissues, or subcellular organelles, followed by purification using density gradient centrifugation and chromatographic techniques (exclusion, affinity, etc.).
- Separation of proteins: Two-dimensional gel electrophoresis is applied for separation of proteins on the basis of their isoelectric points in one dimension and molecular weight in the other. Spots are detected using fluorescent dyes or radioactive probes.
- Identification of proteins: The separated protein spots on the gel are excised and digested in-gel by a protease (e.g., trypsin). The eluted peptides are identified using mass spectrometry.
Analysis of protein molecules is usually carried out by MALDI-TOF (Matrix-Assisted Laser Desorption Ionization–Time of Flight) based peptide mass fingerprinting. The determined amino acid sequence is finally compared with available databases to validate the proteins.
Several online tools are available for proteomic analysis such as Mascot, Aldente, Popitam, Quickmod, Peptide Cutter, etc.
Applications of Proteomics
Proteomics has broad applications in all aspects of life sciences, including practical applications such as drug development against several diseases. Differences in protein expression profiles of normal and diseased individuals may be analyzed to identify target proteins. From protein to gene relationships may be predicted. Once a protein or gene is identified, its function may be predicted. This can help in disease management and drug development.
Whole genome sequences of several organisms have been completed, but genomic data does not show how proteins function or how these proteins are involved in biological processes. A gene codes for a protein, but on several occasions proteins are modified after synthesis through various types of post-translational modifications for functional diversification.
Introduction to Protein Isolation
Protein isolation, also known as protein extraction, is a fundamental biochemical process by which proteins are recovered from cells or tissues for analytical purposes. Isolating proteins is crucial for characterizing their structure and function, studying enzyme mechanisms, analyzing protein interactions, and understanding the molecular basis of various diseases for accurate diagnosis. The complete isolation and purification procedure generally follows five distinct steps: extraction, precipitation, purification, concentration, and storage.
Step 1: Extraction and Cell Disruption
The first stage involves sourcing the protein from materials like microbial cells, plant parts, animal tissues, or food, and obtaining a crude extract. This is achieved by grinding the source material in an appropriate buffer solution under a highly controlled environment, which includes regulating the pH and adding inhibitors to prevent the proteins from denaturing. The specific extraction method chosen depends entirely on the nature of the source material:
- Cryogenic Grinding: This method involves grinding tissues in liquid nitrogen. The extremely low temperature protects the protein structure, making it highly effective for hard tissues like plant roots and stems, or hard-walled cells like cyanobacteria.
- Ultrasonic Homogenisation (Sonication): Utilized primarily for soft tissues, leaves, and microbial cultures, this technique uses an ultrasonic homogenizer to disrupt cells and sub-cellular structures through ultrasonic waves and a phenomenon known as cavitation (the formation of small vapor-filled cavities in liquids).
- Lysis Buffer: A chemical approach suitable for animal and bacterial cells that requires no machinery. A lysis buffer breaks the cell membrane by altering the pH and often contains ionic salts, detergents like sodium dodecyl sulfate (SDS) to solubilize membranes, and chelators like EDTA to sequester metal ions.
- French Press: Designed for resilient microbial plasma membranes and chloroplasts, this device forces a liquid sample through a valve using a hydraulic pump. The resulting shear stress and decompression effectively disrupt the cells.
- Teflon Homogenizer: A mechanical tool consisting of a piston-type pestle and a glass vessel, frequently used in biochemistry labs to prepare homogenates of soft animal tissues such as the liver, kidney, brain, and heart.
Step 2: Centrifugation
Immediately following the grinding or homogenization process, the resulting mixture must be clarified via centrifugation. Centrifugation utilizes centrifugal forces to sediment the mixture components based on their weight and density. The lysized cells suspended in buffer are placed in a centrifuge and spun at high speeds—for example, at 2000xg for 5 to 7 minutes or at 14,000 revolutions per minute (rpm) for 15 minutes, depending on the target cellular material. The centrifugal force drives the heavy cellular debris to the bottom to form a solid “pellet,” which is discarded. The remaining liquid, known as the “supernatant,” contains the desired soluble cytoplasmic proteins and is collected for further processing.
Step 3: Precipitation
Once the supernatant is isolated, precipitation is employed to concentrate the proteins and separate them from the solution, thereby eliminating potential interferences. Different precipitation strategies include acid precipitation, alcohol precipitation, and salting precipitation. In the “salting out” method, adding specific concentrations of salt will cause some proteins to precipitate while others remain dissolved. For example, adding 33-40% saturated ammonium sulphate to a protein mixture will successfully precipitate human serum immunoglobulin, while proteins like albumin will remain entirely in the solution.
Step 4: Advanced Purification Techniques
To isolate the exact protein of interest from the concentrated mixture, advanced purification techniques are utilized based on the protein’s unique physical and chemical properties:
- Separation by Size and Weight: Techniques like ultracentrifugation, gel electrophoresis, and dialysis fall into this category. Dialysis relies on selective, passive diffusion through a semi-permeable membrane to remove unwanted small compounds and salts from the protein solution. Alternatively, Gel-Filtration (Size Exclusion Chromatography) passes the sample through a column packed with porous polymer beads (such as Sephadex or Agarose). Large molecules bypass the pores and elute quickly, while smaller molecules enter the pores and travel through the column much more slowly, allowing for precise size-based separation.
- Separation by Charge: Ion Exchange Chromatography (IEC) separates proteins based on their net electrical charge. The protein mixture is poured through a column packed with a charged matrix—either an anion exchange resin (positively charged) or a cation exchange resin (negatively charged). At a specific pH, the charged proteins bind to the oppositely charged resin groups via reversible electrostatic interactions, allowing targeted proteins to be isolated based on the tightness of their binding.
- Separation by Binding Properties: For highly specific isolation, affinity chromatography or immuno-chromatography utilizes immobilized enzymes and specific binding interactions to capture the exact protein of interest.
Protein Purification and Sequencing Strategies
PART I: Strategies for Protein Purification
The objective of protein purification is to isolate a single protein of interest from a complex biological mixture while preserving its native conformation and biological activity. In modern molecular biology, most proteins are produced recombinantly in host systems such as E. coli, yeast, or mammalian cells. Recombinant expression increases yield and allows the introduction of affinity tags that greatly simplify purification. Regardless of the source, purification follows a logical progression: cell disruption, bulk fractionation, high-resolution chromatographic separation, and polishing steps.
For intracellular proteins, purification begins with cell disruption. Cells may be lysed mechanically using methods such as sonication or French press, or enzymatically using lysozyme (particularly for bacterial cells). Following lysis, the crude extract contains soluble proteins, nucleic acids, membrane fragments, and unbroken cells. Differential centrifugation is therefore performed. Low-speed centrifugation removes large debris and intact cells, while high-speed ultracentrifugation removes ribosomes and membrane vesicles. The lysate often becomes highly viscous due to released DNA and RNA; this is reduced by treatment with DNase, RNase, or polyamines such as polyethyleneimine.
Recombinant proteins frequently accumulate as inclusion bodies, which are dense aggregates of misfolded and insoluble protein. These inclusion bodies are isolated by centrifugation and washed with mild detergents such as Triton X-100 to remove contaminants. They are then solubilized in strong denaturing agents such as 6–8 M guanidine hydrochloride or 8 M urea, often in the presence of a reducing agent like DTT to break incorrect disulfide bonds. The unfolded protein must subsequently be refolded carefully by gradual removal of denaturant and controlled oxidation to restore proper disulfide linkages and native structure.
After extraction, initial fractionation steps are used to concentrate the protein and remove bulk contaminants. One classical method is salting out using ammonium sulfate. Increasing salt concentration reduces protein solubility by competing for water molecules, causing selective precipitation without denaturation. Each protein precipitates at a characteristic salt concentration, enabling partial purification. Another approach involves adjusting the pH to the protein’s isoionic point, where the net charge becomes zero and solubility is minimal, resulting in precipitation. Organic solvents such as cold ethanol or acetone may also be used to selectively precipitate proteins; acetone additionally removes lipids. Neutral polymers such as polyethylene glycol promote precipitation through macromolecular crowding, while synthetic polyelectrolytes bind oppositely charged proteins and coprecipitate them.
High-resolution purification relies on chromatographic techniques, which separate proteins based on distinct physicochemical properties. Ion-exchange chromatography separates proteins according to net charge. Anion exchangers bind negatively charged proteins, whereas cation exchangers bind positively charged proteins. Binding is strongly influenced by the buffer pH relative to the protein’s isoelectric point (pI), and elution is achieved by increasing salt concentration or altering pH. Size-exclusion (gel-filtration) chromatography separates proteins according to molecular size and shape; larger molecules elute first because they are excluded from the pores of the matrix. This technique is commonly used as a final polishing step to remove aggregates and small contaminants. Hydrophobic-interaction chromatography separates proteins based on exposed hydrophobic residues. High salt concentrations enhance hydrophobic interactions, and proteins are eluted by decreasing salt concentration. Affinity chromatography provides the highest specificity by exploiting biological interactions such as antigen–antibody binding, enzyme–substrate recognition, or receptor–ligand interaction.
Recombinant proteins are often engineered with affinity tags to simplify purification. A polyhistidine (His) tag consists of consecutive histidine residues that bind specifically to immobilized metal ions (e.g., Ni²⁺ or Co²⁺) in metal-chelate affinity chromatography. Glutathione S-transferase (GST) is a 26-kDa fusion partner that binds immobilized glutathione. Thioredoxin improves solubility and assists in correct folding. After purification, tags may be removed using site-specific proteases such as thrombin, factor Xa, or enterokinase, or chemically using agents such as cyanogen bromide.
PART II: Protein Sequencing and Characterization
Once purified, a protein must be characterized to confirm that its amino acid sequence matches the predicted cDNA translation and to identify any post-translational modifications.
A classical method of analysis is amino acid composition determination, in which the protein is hydrolyzed under strong acid conditions to release individual amino acids. These are separated and quantified by high-performance liquid chromatography (HPLC). This analysis confirms the relative proportions of amino acids and helps verify overall composition and molecular mass.
For sequence determination, Edman degradation is used to determine the N-terminal amino acid sequence. In this method, phenylisothiocyanate reacts with the N-terminal residue, which is then cleaved and identified without disrupting the remaining peptide chain. The process is repeated sequentially and can determine up to 50–100 residues. However, this method cannot be used if the N-terminus is chemically blocked.
To verify internal sequences, peptide mapping (fingerprinting) is performed. The protein is first denatured, reduced to break disulfide bonds, and alkylated to prevent their reformation. It is then cleaved using specific proteases such as trypsin (which cleaves at lysine and arginine residues), Lys-C, or Asp-N, or chemically using cyanogen bromide, which cleaves at methionine residues. The resulting peptide fragments are separated by HPLC or high-resolution SDS-PAGE. The peptide pattern is compared with predicted fragments to detect sequence errors or structural variations.
Modern protein characterization relies heavily on mass spectrometry (MS). Techniques such as electrospray ionization (ESI-MS) and matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) allow precise determination of molecular mass with very high accuracy. MS can analyze intact proteins or peptide fragments and directly compare measured masses with theoretical values derived from gene sequences. This has largely replaced classical sequencing methods due to its speed, sensitivity, and precision.
Mass spectrometry is also crucial for detecting post-translational modifications (PTMs) such as phosphorylation, oxidation, acetylation, or deamidation. These modifications produce predictable changes in molecular mass. Disulfide bond mapping is performed by digesting the protein without prior reduction, analyzing the fragments by HPLC and MS, then reducing and reanalyzing the sample. Differences in fragment profiles reveal the positions of disulfide linkages.
Finally, computational sequence analysis plays a central role in modern protein characterization. Sequence data are compared against global databases using programs such as BLAST and FASTA to identify homologous proteins and evolutionary relationships. Bioinformatic tools can also predict structural features, including hydrophobic transmembrane regions (hydropathy analysis), secondary structure elements such as alpha helices and beta sheets, and potential functional domains.
Two-Dimensional (2D) Gel Electrophoresis
Two-dimensional (2D) gel electrophoresis is a high-resolution technique used to separate complex mixtures of proteins. It separates proteins based on two independent properties: isoelectric point (pI) in the first dimension and molecular weight (Mr) in the second dimension. Because separation occurs in two perpendicular directions, it provides much higher resolution than one-dimensional SDS-PAGE and is widely used in proteomics.
In the first dimension, proteins are separated by isoelectric focusing (IEF). A pH gradient is established in the gel, and proteins migrate under an electric field until they reach the pH equal to their isoelectric point, where their net charge becomes zero and movement stops. In the second dimension, the focused strip is treated with SDS, which denatures proteins and gives them a uniform negative charge. The proteins are then separated by SDS-PAGE according to molecular weight, with smaller proteins moving faster. Each protein appears as a distinct spot, where horizontal position represents pI and vertical position represents molecular weight.
A major advantage of 2D gel electrophoresis is its ability to detect protein microheterogeneity. Small changes in charge caused by post-translational modifications can shift protein position on the gel. Modifications such as deamidation, phosphorylation, oxidation, carbamylation, or partial proteolytic cleavage may produce multiple closely spaced spots for a single protein.
This technique is widely used for proteome analysis, comparison of protein expression patterns, and evaluation of recombinant protein purity. An advanced form, 2D-DIGE, allows quantitative comparison of different samples in the same gel. After separation, proteins are visualized by staining, excised from the gel, digested with proteolytic enzymes such as trypsin, and analyzed by HPLC or mass spectrometry for identification.
A specialized variation called two-dimensional titration curve analysis combines IEF and perpendicular electrophoresis to study changes in isoelectric point with pH. It is useful for studying protein unfolding, ligand binding, and determining optimal pH conditions for ion-exchange chromatography.
2D gel electrophoresis is a powerful and sensitive technique that separates proteins based on charge and size, detects structural variations, and plays a central role in protein characterization and proteomic research.
Introduction to Chromatography
Chromatography is a versatile physical method of separation in which the components of a mixture are separated by their distribution between two phases. One phase remains stationary (the stationary phase), while the other phase (the mobile phase) moves through it in a definite direction. The term itself is derived from two words: “Chromo,” meaning color, and “Graphy,” meaning the representation of something on paper. The technique was invented in 1901 by the Russian botanist Mikhail Tswett during his research on plant pigments. He successfully used a glass column filled with powdered limestone as his stationary phase and ethanol as his mobile phase to separate pigments like chlorophylls, xanthophylls, and carotenoids.
Principle of Operation
The fundamental principle of chromatography relies on the continuous physical equilibrium and the differing affinities that sample components have for the stationary and mobile phases. The sample is transported by the mobile phase—which can be a gas, liquid, or supercritical fluid—and is forced through a stationary phase held in a column or on a solid surface. As the sample moves, its components interact with the stationary phase. Substances that interact strongly (high affinity) with the stationary phase are retained longer and therefore move more slowly through the system. Conversely, substances that interact weakly (low affinity) move much more quickly. This difference in travel rates ultimately causes the mixture to separate into its individual discrete components.
Key Terminologies and Mathematical Concepts
Several important technical terms and equations are used to define the chromatographic process. Retention time ( ) is the characteristic time it takes for a particular analyte to pass completely through the system, from the injection inlet to the detector. The equilibrium of a solute between the two phases is described by the distribution coefficient (
) is the characteristic time it takes for a particular analyte to pass completely through the system, from the injection inlet to the detector. The equilibrium of a solute between the two phases is described by the distribution coefficient (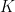 ), which is calculated as
), which is calculated as  , where
, where  is the molar concentration of the solute in the stationary phase and
is the molar concentration of the solute in the stationary phase and  is the concentration in the mobile phase. Furthermore, the efficiency of a chromatographic column is measured by the number of theoretical plates (
is the concentration in the mobile phase. Furthermore, the efficiency of a chromatographic column is measured by the number of theoretical plates (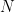 ), which can be calculated using the formula
), which can be calculated using the formula  , where
, where  represents the width of the peak’s base. Finally, in planar chromatography, the retardation factor (
represents the width of the peak’s base. Finally, in planar chromatography, the retardation factor (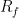 ) is used to identify components, defined as the distance moved by the substance divided by the distance moved by the solvent front.
) is used to identify components, defined as the distance moved by the substance divided by the distance moved by the solvent front.
Major Types of Chromatography
There are several highly specialized types of chromatography tailored to different types of samples.
- Gas Chromatography (GC): Separates vaporized samples utilizing a carrier gas (like nitrogen or helium) as the mobile phase and is specifically highly effective for volatile samples up to about 250°C.
- Liquid Chromatography (LC) & HPLC: Liquid chromatography separates liquid samples using a liquid solvent mobile phase. High-Performance Liquid Chromatography (HPLC) is an advanced, widely used analytical technique where a sample is injected into a column packed with very small particles under high pressure, making it highly useful for pharmaceuticals and biomolecules.
- Thin-Layer Chromatography (TLC): Uses a liquid solvent as the mobile phase and a glass plate covered with a thin layer of alumina or silica gel as the stationary phase to separate dried liquid samples.
- Specialized Techniques: Gel filtration separates molecules according to their differences in physical size, which is highly suited for sensitive biomolecules. Ion exchange chromatography separates components based on reversible competitive binding of ions, while affinity chromatography relies on the specific complex formation between pairs of biomolecules.
Core Applications
Chromatography is a vital analytical tool used extensively across scientific and industrial disciplines to separate, analyze, identify, purify, and quantify mixtures. It is widely implemented in pharmaceutical companies, environmental agencies for tracking trace ions in pollution, and hospitals for clinical analysis. In biochemistry, it is indispensable for the bioseparation of complex molecules such as DNA, carbohydrates, lipids, proteins, and amino acids.
Protein sequencing by Edman degradation
The sequence of amino acids in a protein or peptide can be identified by Edman degradation, which was developed by Pehr Edman.
This method can label and cleave the peptide from N-terminal without disrupting the peptide bonds between other amino acid residues.
The Edman degradation reaction was automated in 1967 by Edman and Beggs.
Nowadays, the automated Edman degradation (the protein sequenator) is used widely, and it can sequence peptides up to 50 amino acids i.e. small peptides can be sequenced. For larger peptides, we need to break them into small peptides and then sequence them separately. These separate sequences are then used to reconstruct the sequence of the larger protein but discussion of these methods for larger proteins is out of scope here.
This process is used to confirm the identity of the recombinant proteins after their production, isolation and purification. This is done by sequencing the N-terminal amino acid residues.
Stepwise mechanism of Edman’s degradation:
- Peptide is made to react with phenylisothiocyanate (PITC) at the amino terminus under mildly alkaline conditions to give a phenylthiocarbamoyl derivative (PTC-peptide).
- Then, conditions are changed to acidic by adding anhydrous trifluoroacetic acid (CF3COOH).
- This leads to the thiocarbonyl sulfur of the PTC-derivative attacking the carbonyl carbon of the N-terminal amino acid. Thus, peptide bond linking the N-terminal amino acid to the rest of the peptide is attacked for cleavage.
- As a result of this reaction, first amino acid is cleaved as anilinothiazolinone derivative (ATZ-amino acid) and the remaining peptide is isolated and subjected to the next degradation cycle.
- Once formed, the ATZ-derivative (also called as thiazolinone derivative) is more stable than phenylthiocarbamoyl derivative.
- The ATZ amino acid is now separated by extraction with ethyl acetate and converted to a phenylthiohydantoin derivative (PTH-amino acid) by treatment with aqueous acid.
- Chromatography is then used to identify the PTH residue generated by each cycle.

Mass Spectrometry: Principle, Instrumentation, Working and Applications
Mass spectrometry (MS) is an advanced analytical technique used to determine the mass-to-charge ratio (m/z) of ions for the identification and quantification of chemical substances. The method works by converting molecules into ions and analyzing their motion in electric or magnetic fields. Through this process, it provides precise information about molecular mass, isotopic distribution, and structural characteristics. The final output, known as a mass spectrum, is a graphical representation of relative ion intensity versus m/z values and serves as a molecular fingerprint for compound identification.
Principle of Mass Spectrometry
The core principle of mass spectrometry is based on the formation of gas-phase ions from sample molecules followed by their separation according to mass-to-charge ratio.
Typically, molecules are ionized by exposure to high-energy electrons, which remove one or more electrons to generate positively charged ions (commonly singly charged, +1). During this process, the molecular ion may remain intact or fragment into smaller ions. Each resulting ion possesses a specific m/z value. For singly charged species, the m/z value closely corresponds to the ion’s molecular mass.
After ionization, the ions are accelerated so that they acquire uniform kinetic energy. They are then directed through electric or magnetic fields, where separation occurs based on differences in m/z. Ions with lower mass or higher charge experience greater deflection compared to heavier or lower-charge ions. A detector measures the ions as they arrive, and the collected data generate a mass spectrum. The position of each peak indicates its m/z value, while the peak intensity reflects relative abundance. Structural interpretation is achieved by analyzing characteristic fragmentation patterns and comparing results with reference databases.
Instrumentation and Steps in Mass Spectrometry
A mass spectrometer operates under high vacuum conditions to minimize collisions between ions and gas molecules. The essential components function sequentially as follows:
Sample Inlet
The sample—whether solid, liquid, or gas—is introduced into the instrument. It enters the ionization chamber at low pressure through a controlled system such as a molecular leak inlet or direct insertion probe.
Ionization Source
Within the ionization chamber, molecules are converted into ions. Common ionization methods include:
- Electron Ionization (EI) — A hard ionization technique involving electron bombardment that often produces extensive fragmentation.
- Chemical Ionization (CI) — A softer method that reduces fragmentation and is useful for determining molecular mass.
- Fast Atom Bombardment (FAB) — Applied particularly for larger or less volatile molecules.
Acceleration
The positively charged ions pass through a series of accelerating plates or slits maintained at decreasing electrical potentials. This ensures that all ions gain similar kinetic energy before entering the mass analyzer.
Mass Analyzer (Separation System)
The mass analyzer separates ions according to their m/z values. Different analyzer designs include:
- Magnetic Sector Analyzer — Uses a magnetic field to deflect ions along curved trajectories.
- Quadrupole Analyzer — Employs oscillating radiofrequency fields to selectively filter ions.
- Time-of-Flight (TOF) Analyzer — Separates ions based on differences in flight time over a fixed distance.
- Ion Trap Analyzer — Traps ions in electromagnetic fields and sequentially ejects them for detection.
Detector
The separated ions strike a detector, such as an electron multiplier. Upon impact, the ions are neutralized and generate an amplified electrical signal proportional to their abundance. This signal is processed electronically to produce the mass spectrum.
Working of Mass Spectrometry
In practical operation, the sample is introduced and vaporized if necessary. Ionization generates a mixture of molecular ions and fragment ions. All ions are accelerated to comparable kinetic energies before entering the mass analyzer, where they are separated according to m/z-dependent motion or flight times.
As ions reach the detector, their arrival is recorded and translated into a spectrum. Interpretation of the mass spectrum involves identifying the molecular ion peak, which corresponds to the intact molecule’s mass, and analyzing fragment peaks to deduce structural features. Comparison with established spectral libraries enhances compound identification accuracy.
Applications of Mass Spectrometry
Mass spectrometry is widely applied across scientific and industrial fields due to its high sensitivity, specificity, and ability to analyze complex mixtures.
- Used for detection of environmental contaminants such as pesticides, industrial pollutants, and trace organic compounds in air, water, and soil. It is also applied in monitoring food safety.
- Facilitates isotopic analysis, age determination of rocks, and exploration of petroleum and natural gas reserves.
- Applied in quality control, purity assessment, and structural verification of synthetic and industrial chemicals.
- Essential for determining accurate molecular masses of peptides, proteins, nucleic acids, and oligonucleotides. It supports protein sequencing, post-translational modification analysis, and structural characterization of carbohydrates and other biomolecules.
- Used in detecting drugs of abuse, toxicological screening, monitoring metabolites in biological fluids, and analyzing anesthetic or respiratory gases during surgery.
- Supports drug discovery, metabolomics, aerosol particle analysis, and advanced proteomics research.
Notes on Endopeptidases
Endopeptidases (also called endoproteinases) are a major class of proteolytic enzymes (proteases or peptidases) that catalyze the hydrolysis of peptide bonds within the interior of polypeptide chains or proteins. Unlike exopeptidases, which cleave peptide bonds at the terminal ends (N- or C-terminus) releasing individual amino acids or small peptides, endopeptidases act on non-terminal peptide bonds, breaking long protein chains into smaller polypeptide fragments. This internal cleavage increases the number of free ends available for further digestion by exopeptidases, playing a crucial role in protein degradation and digestion.
Differences Between Endopeptidases and Exopeptidases
- Site of action — Endopeptidases cleave internal peptide bonds (away from termini), producing shorter peptides; exopeptidases cleave terminal peptide bonds, releasing amino acids one by one.
- Products — Endopeptidases generate oligopeptides and polypeptides; exopeptidases produce free amino acids or dipeptides.
- Role in digestion — Endopeptidases initiate breakdown by fragmenting large proteins; exopeptidases complete digestion into absorbable units.
- Specificity — Endopeptidases are often highly specific for certain amino acid residues flanking the cleaved bond; exopeptidases target the terminal residue type.
Classification of Endopeptidases
Endopeptidases are classified based on their catalytic mechanism (nature of the active site residues or cofactors involved in hydrolysis):
- Serine endopeptidases (EC 3.4.21) — Use a serine residue in the catalytic triad (serine, histidine, aspartate); most common in digestion (e.g., trypsin, chymotrypsin, elastase).
- Cysteine endopeptidases (EC 3.4.22) — Depend on a cysteine residue for nucleophilic attack (e.g., papain, cathepsins).
- Aspartic endopeptidases (EC 3.4.23) — Use two aspartic acid residues activated by acid-base catalysis (e.g., pepsin, renin).
- Metallo-endopeptidases (EC 3.4.24) — Require metal ions (usually Zn²⁺) for catalysis (e.g., thermolysin, matrix metalloproteinases).
- Threonine endopeptidases (EC 3.4.25) — Use threonine as the nucleophile (e.g., proteasome subunits).
- Glutamic endopeptidases — Less common, involve glutamic acid in catalysis.
Major Examples of Endopeptidases (Focus on Digestive Ones)
In mammalian protein digestion, key endopeptidases include:
- Pepsin (aspartic endopeptidase) — Secreted in the stomach as pepsinogen (zymogen), activated by low pH (HCl); cleaves bonds involving aromatic amino acids (Phe, Tyr, Trp) and others (Leu, Met); optimal at pH 1.5–2.5; initiates protein digestion in acidic environment.
- Trypsin (serine endopeptidase) — Produced in pancreas as trypsinogen, activated by enterokinase (duodenum); highly specific, cleaves after basic residues (Lys, Arg) on the carboxyl side (not followed by Pro); optimal at pH ~8.
- Chymotrypsin (serine endopeptidase) — From chymotrypsinogen (activated by trypsin); cleaves after large hydrophobic/aromatic residues (Phe, Tyr, Trp, Met, Leu); optimal at pH ~8.
- Elastase (serine endopeptidase) — From proelastase (activated by trypsin); cleaves after small neutral residues (Ala, Gly, Ser, Val); helps digest elastin and other proteins.
These pancreatic enzymes work synergistically in the small intestine to fragment polypeptides into oligopeptides.
Functions of Endopeptidases
- Protein digestion — Break dietary proteins into smaller peptides in the gastrointestinal tract, facilitating complete hydrolysis to amino acids.
- Protein processing — Activate zymogens, cleave pro-proteins (e.g., insulin maturation), and regulate biological pathways.
- Cellular processes — Involved in apoptosis (caspases, cysteine endopeptidases), antigen processing (proteasome, threonine endopeptidases), extracellular matrix remodeling (metalloproteinases), and pathogen defense.
- Industrial/biotechnological uses — Food processing (e.g., tenderizing meat, cheese production), detergents, and peptide synthesis.
Mechanism of Action (General, with Serine Protease Example)
Most digestive endopeptidases (serine type) follow the catalytic triad mechanism:
- The substrate binds to the enzyme’s active site, positioning the target peptide bond.
- Nucleophilic attack Serine’s hydroxyl group (activated by histidine and aspartate) attacks the carbonyl carbon of the peptide bond, forming a tetrahedral intermediate.
- Acyl-enzyme intermediate The peptide bond breaks, releasing the C-terminal fragment; the N-terminal part remains covalently attached to serine.
- Deacylation Water (activated by histidine) hydrolyzes the acyl-enzyme, releasing the N-terminal fragment and regenerating the enzyme.
This mechanism ensures precise, efficient cleavage at specific sites.
Endopeptidases are essential for efficient protein turnover and nutrient absorption.
Serial Analysis of Gene Expression (SAGE)
Serial Analysis of Gene Expression (SAGE) is a sequencing-based transcriptomic technique that enables global, quantitative, and unbiased profiling of gene expression by generating short sequence tags from mRNA transcripts. Developed in 1995 by Velculescu et al., SAGE provides a digital snapshot of the transcriptome, allowing simultaneous analysis of thousands of transcripts without prior knowledge of gene sequences. It is particularly useful for discovering novel genes and comparing expression profiles between samples (e.g., normal vs. diseased tissues).
Fundamental Principles of SAGE
SAGE is based on two core principles:
- A short oligonucleotide sequence (tag, typically 9–14 bp) from the 3′-end of each mRNA, defined by a specific restriction site at a fixed distance from the poly(A) tail, is sufficient to uniquely identify the transcript. A 10-bp tag theoretically yields 4¹⁰ = 1,048,576 unique combinations—adequate to distinguish most transcripts in complex genomes like humans.
- Concatenation of multiple tags into long serial chains (concatemers) enables efficient, high-throughput sequencing of many tags in a single reaction, maximizing data output per sequencing run.
Methodology and Step-by-Step Protocol
The SAGE protocol is multi-step and labor-intensive, involving enzymatic digestion, ligation, amplification, and cloning under strict conditions to minimize bias.
- mRNA Isolation and cDNA Synthesis
- Extract total RNA and isolate poly(A)+ mRNA using biotinylated oligo(dT) primers.
- Synthesize double-stranded cDNA directly on streptavidin-coated magnetic beads (biotin binds to streptavidin).
- Anchoring Enzyme Digestion
- Digest cDNA with an anchoring enzyme (typically NlaIII, a 4-cutter restriction endonuclease) that cleaves at the 3′-most recognition site, leaving the tag region anchored to the bead.
- Linker Ligation and Division
- Divide the bead-bound cDNA into two aliquots.
- Ligate different linker oligonucleotides (Linker A and Linker B) to each aliquot. These linkers contain recognition sites for a Type IIS restriction enzyme (tagging enzyme).
- Tag Release (Tagging Enzyme Digestion)
- Use the tagging enzyme (BsmFI in standard SAGE) to cleave 10 bp downstream of its recognition site, releasing short SAGE tags (≈10–14 bp) from the beads.
- Ditag Formation
- Blunt-end the released tags.
- Ligate tags from the two pools to form ditags (paired tags joined head-to-tail).
- PCR Amplification
- Amplify ditags using primers specific to the linkers.
- Concatemer Construction
- Cleave amplified ditags to remove linkers.
- Gel-purify and serially ligate ditags to form long concatemers (chains of 20–50+ ditags).
- Cloning and Sequencing
- Clone concatemers into a plasmid vector.
- Transform into bacteria for amplification.
- Sequence clones using automated Sanger sequencing to obtain tag sequences.
Data Analysis
- Use specialized software (e.g., SAGE2000 or modern equivalents) to:
- Extract individual tags from sequencing reads.
- Remove duplicates and artifacts.
- Quantify tag abundance (digital count directly reflects transcript level).
- Map tags to genes using reference databases (e.g., SAGEmap from NCBI, UniGene, or RefSeq).
- Compare tag frequencies between libraries to identify differentially expressed genes.
Modifications and Variants
- LongSAGE: Uses MmeI as tagging enzyme to generate longer 17-bp tags → improves unique gene mapping and annotation accuracy.
- SuperSAGE / RL-SAGE: Produce even longer tags (up to 26 bp) for better specificity.
- MicroSAGE / SAGE-Lite: Require much less starting mRNA (nanogram quantities) → suitable for limited samples (e.g., biopsies, rare cells).
- These variants address limitations of standard SAGE and enhance resolution.
Advantages of SAGE
- Digital and quantitative: Tag counts provide absolute expression levels (no hybridization bias).
- Unbiased and open-ended: No need for prior gene sequence knowledge → discovers novel/unknown transcripts.
- Highly reproducible: Direct sequencing avoids probe-related variability seen in microarrays.
- Sensitive to low-abundance transcripts with sufficient sequencing depth.
Limitations of SAGE
- Labor-intensive and time-consuming (multiple enzymatic steps, cloning required).
- Short tags (9–14 bp in standard SAGE) may lead to tag-to-gene ambiguity (multiple genes sharing identical tags) or failure to identify unknown genes.
- Restriction enzyme dependence → potential biases if sites absent or multiple per transcript; species-specific incompatibilities.
- Low throughput compared to modern NGS-based methods (e.g., RNA-seq).
- Requires large amounts of starting material in original protocol (though variants mitigate this).
Applications
- Global gene expression profiling in normal vs. diseased states (e.g., cancer vs. normal tissue).
- Discovery of novel biomarkers (e.g., identification of prostate stem cell antigen (PSCA) as a pancreatic cancer marker via SAGE database comparison).
- Transcriptome analysis in non-model organisms or poorly annotated genomes.
- Differential expression studies in development, stress response, or drug treatment.
- Complements other techniques like microarrays or RNA-seq for validation.
Mechanism Of Gene Prediction
The fundamental challenges in eukaryotic gene prediction stem from the complex structure of eukaryotic genomes compared to prokaryotes. The two basic problems in gene prediction are identifying protein-coding regions and predicting the functional sites of genes. This is complicated in eukaryotes by low gene density and very large intergenic spaces that contain multiple repeated sequences and transposable elements. Eukaryotic genes are “split,” meaning they contain both coding exons and non-coding introns. Furthermore, predicting these structures must account for complex biological processes: transcripts are capped via the methylation of the 5′ residue, splicing occurs in the spliceosome, and alternative splicing can occur.
A major issue is the identification of exact splicing sites, which generally follow the GT-AG rule (specifically, the GTAAGT / Y12NCAG 5’/3’ intron splice junctions). Another complication is polyadenylation, which involves approximately 250 adenine nucleotides being added downstream of the CAATAAA(T/C) consensus box.
To address these complexities, computational gene prediction programs have evolved through four distinct generations:
- First Generation: Programs like TestCode and GRAIL were limited to identifying the approximate locations of coding regions within genomic DNA.
- Second Generation: Programs like SORFIND and Xpound advanced by combining splice signal and coding region identification to predict potential exons; however, they did not attempt to assemble these predicted exons into complete genes.
- Third Generation: Programs such as GeneParser, GenLang, and FGENEH attempted to predict complete gene structures. Their performance was often poor because they relied on the flawed assumption that the input sequence contained exactly one complete gene.
- Fourth Generation: Modern programs like GENSCAN, AUGUSTUS, and GENEID were developed to solve the “one complete gene” limitation, significantly improving accuracy and applicability for real-world genomic sequences.
The mechanisms utilized by these programs generally fall into two categories:
1. Sequence Similarity-Based (Extrinsic) Methods This extrinsic approach identifies genes by conducting homology searches against known databases, such as genomic DNA, dbEST, or protein databases. Comparing two homologous genomic sequences facilitates the identification of conserved exons. To accurately model the gene structure, organization, and refine region boundaries, this method is combined with signal sensors that detect biological signals relating to transcription, translation, and splicing.
2. Ab Initio-Based (Intrinsic) Methods This method relies solely on the provided sequence without any prior information about the gene. It predicts structure using signal and content sensors, such as Poly-A sites and intron splice sites. Intrinsic methods frequently use nucleotide composition-based mathematics, most notably Hidden Markov Models (HMM). An HMM assumes that the probability of a given nucleotide occurring is dependent on the previous k nucleotides by applying conditional probabilities. The Generalized Hidden Markov Model (GHMM) is the most commonly used framework because it allows an entire string of sequence to be an output of a given state.
Gene Prediction Tools:
- GENSCAN: Developed by Chris Burge at Stanford University’s Department of Mathematics, GENSCAN is a general-purpose program that analyzes genomic DNA from humans, other vertebrates, invertebrates, and plants. It relies on a GHMM (General Hidden Markov Model library) to predict the location of genes and their exon-intron boundaries. It accepts sequence files in either FASTA or minimal GenBank formats.
- AUGUSTUS: Also based heavily on a Generalized Hidden Markov Model (GHMM), this program is specifically designed to predict genes in eukaryotic genomic sequences and can be run through a web interface.
- GENEID: This program is designed with a highly efficient hierarchical structure and processes anonymous genomic sequences. It is incredibly fast, with version 1.2 capable of analyzing 1Gbp per hour (processing the whole human genome in 3 hours). Its mechanism operates in three distinct steps:
- Splice sites, start codons, and stop codons are predicted and scored along the sequence using Position Weight Arrays (PWAs).
- Exons are built from these sites. They are scored by adding the sum of the scores of the defining sites to the log-likelihood ratio of a Markov Model specifically for coding DNA.
- The final gene structure is assembled from the set of predicted exons by maximizing the total sum of the scores of the assembled exons.
- GENIE: This system uses a GHMM that incorporates specialized signal and content sensors. The most extensively studied model within Genie is the sensor used to predict coding regions (exons). These content sensors are heavily based on coding usage, coding preferences, and expected length distributions. While initially optimized exclusively for human genes, the current Genie system has been newly trained to include optimizations for Drosophila melanogaster.
- EUGENE: Functioning as an “open integrative gene finder” for both eukaryotic and prokaryotic genomes, EuGene is unique in its ability to integrate arbitrary, external sources of information into its prediction process. This includes data from RNA-Seq, protein similarities, homologies, and various statistical sources. Its Eukaryote Pipeline (EuGene-EP) exploits probabilistic mathematical models, like Markov models, to discriminate coding from non-coding sequences and to mathematically differentiate effective, real splice sites from false ones.
Protein – Protein Interaction (PPI) Databases and Tools
Protein–protein interactions (PPIs) are fundamental to cellular organization and biological function. Virtually every cellular process signal transduction, transcriptional regulation, immune response, cytoskeletal organization, apoptosis, cell cycle control, and metabolic coordination—depends on precise physical or functional interactions between proteins.
The Role of Proteins in Systems Biology
In systems biology, proteins rarely act in isolation; instead, they operate within highly interconnected interaction networks. Studying PPIs enables researchers to reconstruct cellular pathways, identify disease-associated perturbations, discover biomarkers, and design therapeutic targets. With the rapid expansion of high-throughput technologies such as yeast two-hybrid screening and affinity purification–mass spectrometry (AP-MS), large-scale interaction datasets have accumulated, leading to the development of specialized PPI databases and analytical tools.
Classification of PPI Databases
PPI databases are broadly categorized into primary databases and secondary (meta) databases. Primary databases contain experimentally validated interaction data curated directly from peer-reviewed literature. These databases emphasize accuracy, standardized annotation, and experimental detail. Secondary databases integrate data from multiple primary resources and often incorporate computational predictions, text mining, and functional associations. This distinction is crucial: primary databases focus on experimentally supported interactions, whereas secondary databases combine experimental and predicted data to provide broader network coverage.
Key Primary Databases
IntAct is one of the most important primary databases, maintained by EMBL-EBI. It provides manually curated molecular interaction data extracted from scientific publications and follows standardized formats such as PSI-MI (Proteomics Standards Initiative–Molecular Interaction). IntAct stores detailed metadata including experimental methods, detection techniques, host organisms, and interaction types. It assigns confidence measures and allows network visualization, making it a gold-standard curated repository. Due to its strict curation policy and standardized vocabulary, IntAct is frequently referenced as a reliable experimental PPI resource.
DIP (Database of Interacting Proteins) is one of the earliest PPI repositories. It focuses on experimentally determined protein interactions and provides curated interaction maps. DIP emphasizes high-quality binary interactions derived from controlled experiments. Although smaller compared to modern databases, DIP remains historically significant in proteomics and is often cited when discussing the evolution of interaction databases.
BioGRID (Biological General Repository for Interaction Datasets) is another extensively used primary database. It includes both physical protein–protein interactions and genetic interactions. Physical interactions refer to direct binding between proteins, while genetic interactions describe functional relationships inferred from phenotypic effects. BioGRID covers multiple organisms and is frequently updated, making it a comprehensive resource for interaction mapping in model organisms and humans.
MINT (Molecular INTeraction database) focuses primarily on experimentally verified molecular interactions with manual curation and standardized annotation. It shares conceptual similarities with IntAct and emphasizes accuracy and experimental detail.
HPRD (Human Protein Reference Database) specifically concentrates on human proteins and includes additional biological annotations such as domain architecture, post-translational modifications, tissue expression patterns, and disease associations. HPRD is particularly relevant for studies of human disorders, including cancer and inherited diseases.
Secondary and Integrative Databases
STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) represents a prominent secondary database that integrates experimentally validated data with computational predictions. STRING compiles interaction evidence from diverse sources including experimental data, co-expression analysis, gene neighborhood conservation, gene fusion events, phylogenetic co-occurrence, curated pathway databases, and automated text mining. It distinguishes between direct physical interactions and indirect functional associations. STRING assigns confidence scores (low, medium, high) based on evidence strength and provides enrichment analysis tools for Gene Ontology terms and pathways. It is often highlighted for its integrative nature and scoring system.
Experimental Methods for Detecting PPIs
The experimental methods underlying PPI data are central to understanding database content. Yeast two-hybrid (Y2H) screening detects binary protein interactions by reconstituting a transcription factor in yeast; it is high-throughput but prone to false positives. Co-immunoprecipitation (Co-IP) detects physical interactions in near-physiological conditions and provides stronger biological relevance. Affinity purification–mass spectrometry (AP-MS) identifies protein complexes rather than just binary interactions, making it suitable for studying multiprotein assemblies. Additional methods include protein microarrays, fluorescence resonance energy transfer (FRET), bimolecular fluorescence complementation (BiFC), and cross-linking mass spectrometry. Each technique varies in sensitivity, specificity, throughput, and false discovery rate—an important point for comparative discussions.
Computational Tools for PPI Analysis
Beyond databases, several computational tools assist in analyzing and visualizing PPI networks. Cytoscape is a widely used open-source platform for network construction and analysis. In Cytoscape, proteins are represented as nodes and interactions as edges. The software supports topological analysis such as degree centrality, betweenness centrality, clustering coefficient, and module detection through plugins. It is indispensable in systems biology and network pharmacology studies. GeneMANIA predicts gene function by integrating PPI data with co-expression, co-localization, and pathway information. ClusPro is a structural bioinformatics tool used for protein–protein docking, predicting three-dimensional complex formation based on physicochemical complementarity.
Hub Proteins in PPI Networks
An important conceptual topic in PPI network analysis is the idea of hub proteins. Hub proteins possess a high degree of connectivity within interaction networks and are often essential genes. Biological networks typically exhibit scale-free properties, meaning that most proteins have few interactions while a small number serve as highly connected hubs. Hub proteins frequently participate in critical pathways and are attractive drug targets; however, targeting them may also cause toxicity due to their central cellular roles.
Confidence Scoring in PPI Data
Confidence scoring is a crucial aspect of PPI databases. Interaction reliability is assessed based on experimental method quality, reproducibility, number of supporting publications, evolutionary conservation, and computational evidence strength. High-confidence interactions are supported by multiple independent methods and publications, whereas low-confidence interactions may rely solely on predictive models. Understanding scoring metrics is important for interpreting network robustness and avoiding false-positive-driven conclusions.
Applications of PPI Databases
Applications of PPI databases extend across biomedical research. In disease biology, altered interaction networks help explain cancer progression, neurodegeneration, and immune dysregulation. In drug discovery, network pharmacology approaches identify hub proteins or bottleneck nodes as therapeutic targets. In systems biology, PPI data enable pathway reconstruction and functional annotation of uncharacterized proteins. Comparative interactomics also allows evolutionary analysis of conserved interaction modules across species.
Limitations of PPI Databases
Despite their utility, PPI databases have limitations. False positives and false negatives remain significant challenges, particularly in high-throughput experiments. Data bias toward well-studied proteins leads to incomplete interactomes for less-characterized organisms. Species-specific interactions may not extrapolate across taxa. Furthermore, static network representations often fail to capture temporal dynamics, post-translational modifications, or context-dependent interactions occurring under specific cellular conditions.
PPI databases form the backbone of modern proteomics and systems biology research. Primary databases such as IntAct, DIP, BioGRID, MINT, and HPRD emphasize experimentally validated data, whereas STRING integrates both experimental and predicted interactions. Analytical tools such as Cytoscape, GeneMANIA, and ClusPro facilitate visualization, functional prediction, and structural modeling. Understanding database classification, experimental methods, confidence scoring, hub proteins, and network properties is essential for examination preparation as well as for advanced research applications.














